
FDA AI規制の最新動向と医療機器開発への影響:ガイダンス草案から読み解く開発戦略
FDA(米国食品医薬品局)のAI規制が2025年に大きな転換点を迎える中、医療機器開発に携わる企業の皆様は「FDA AI規制の最新動向を把握できているだろうか?」「新しいガイダンス草案が自社の開発戦略にどのような影響を与えるのか?」といった不安を抱えていませんか?
実際に、2025年1月に発表された「AI-Enabled Device Software Functions」ガイダンス草案により、AI医療機器の透明性要求や継続的学習システムの管理方法が大幅に変更されました。この変化に対応できない企業は、承認取得の遅延や市場競争力の低下といった深刻なリスクに直面する可能性があります。
しかし、適切な対応策を講じることで、これらの規制変更を競争優位に転換することも可能です。本記事では、FDA AI規制の専門知識を持つAIコンサルタントの視点から、2025年ガイダンス草案の詳細分析、SaMD開発における実践的アプローチ、そして成功企業の具体的事例まで、開発戦略立案に必要な情報を包括的に解説します。
FDA AI transparencyの要求事項からPCCP活用による効率化手法まで、今すぐ知っておくべき最新情報が満載です。ぜひ最後までお読みいただき、貴社の医療機器開発戦略にお役立てください。
- FDA AI最新ガイダンスの要点:2025年1月発表のAI医療機器規制の変更点
- PCCP活用による効率化:事前変更計画による迅速な承認取得方法
- AI透明性要求への対応:モデルカード作成と文書化の実践方法
- 成功企業の実例:Amgen社などの具体的なAI活用戦略

FDA AIガイダンスの最新動向と開発への影響
2025年1月に公表されたAI関連ガイダンス草案の概要
FDAは2025年1月に「AI-Enabled Device Software Functions」に関するガイダンス草案を発表し、AI医療機器開発における新たな規制枠組みを明確化しました。
このガイダンスでは、AI搭載医療機器の市販前申請に必要な文書化要件が詳細に規定されており、特にモデルカードの作成や透明性の確保が重要視されています。
従来の医療機器規制とは異なり、AIの継続的学習能力や適応性を考慮した新しい評価基準が導入されています。
AI搭載医療機器ソフトウェア機能のライフサイクルマネジメント
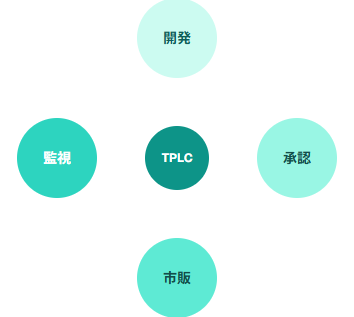
FDAが提唱するTotal Product Lifecycle(TPLC)アプローチでは、AI医療機器の開発から市販後監視まで一貫した品質管理体制の構築が求められています。
このアプローチにより、AI医療機器の性能変化や学習データの更新に対する継続的な監視が可能となります。特に、機械学習アルゴリズムの予期しない動作や性能劣化を早期に検出するためのモニタリングシステムの実装が必須要件として位置づけられています。
医薬品・生物製剤の規制判断を支援するAI活用の考慮事項
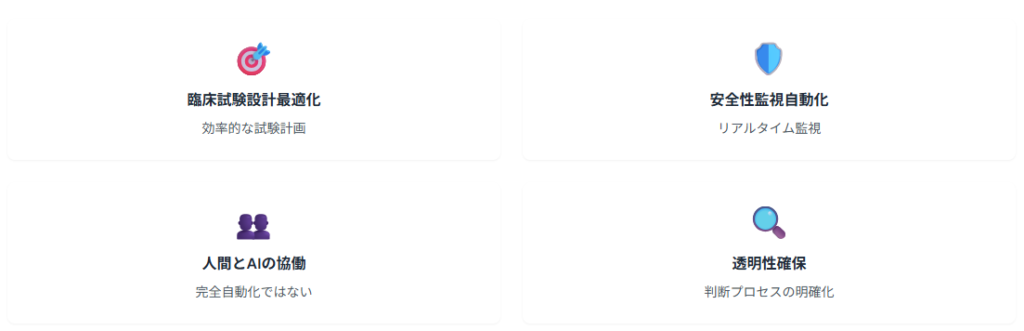
2025年1月にFDAが発行した「Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products」では、医薬品開発プロセスにおけるAI活用の具体的指針が示されています。このガイダンスによると、臨床試験設計の最適化や安全性監視の自動化において、AIの活用が積極的に推奨されています。
ただし、AI判断の透明性と人間による監督体制の確保が前提条件として明記されており、完全自動化ではなく人間とAIの協働モデルが重視されています。
FDAによるAI規制の発展経緯と現在の位置づけ
2023年から2025年にかけての規制動向の変化
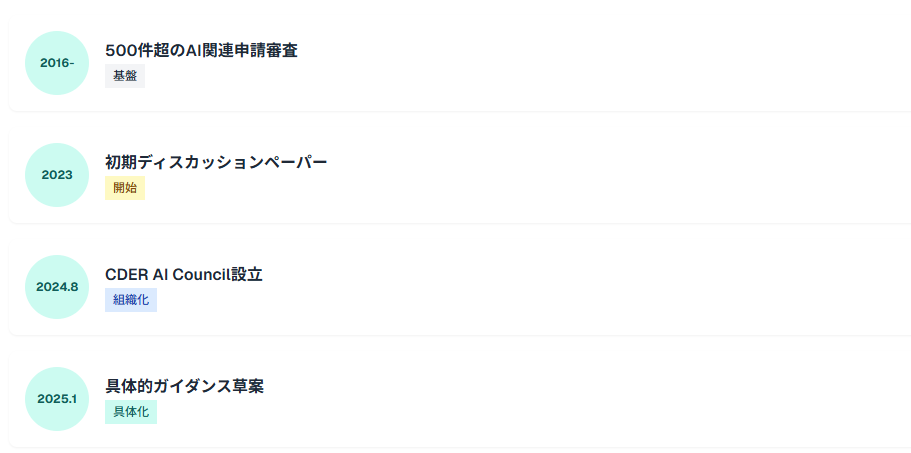
FDAのAI規制は2023年の初期的なディスカッションペーパーから、2025年の具体的なガイダンス草案まで急速に発展しています。
FDAは2016年以降、500件を超えるAI関連申請を審査しており、この申請件数の増加傾向は2025年も継続しています。この申請件数の増加に対応するため、FDAは2024年8月にCDER AI Councilを設立し、組織横断的なAI規制体制を構築しました。
AI/ML技術の医療分野への急速な普及と課題

医療分野におけるAI技術の普及は、診断支援から治療最適化まで幅広い領域に及んでいます。しかし、AIの「ブラックボックス」問題や学習データのバイアス、サイバーセキュリティリスクなど、従来の医療機器にはない新たな課題も浮上しています。
FDAはこれらの課題に対処するため、リスクベースアプローチを採用し、AI技術の利益とリスクのバランスを慎重に評価する方針を示しています。
 ReAlice株式会社 開発担当者
ReAlice株式会社 開発担当者モデルカードやライフサイクル監視の義務化は、単なるアルゴリズム精度ではなく継続的説明可能性と運用監視体制を重視する流れの象徴です。AIを用いた判断の透明性や人間の介在も明記されており、完全自律ではなく共創型AIが中心になる点が今後の方向性を示しています。
FDA AI医療機器開発における透明性と信頼性の要求事項
AIモデルの透明性確保のための具体的要件
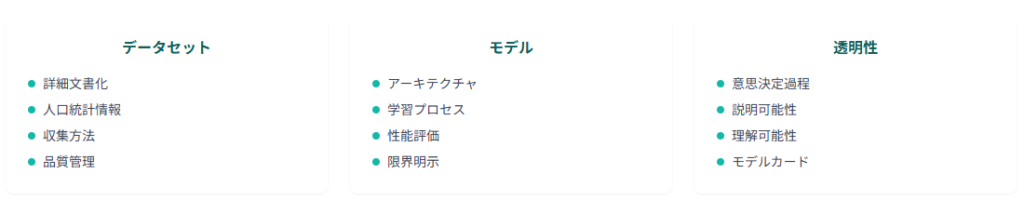
FDAの最新ガイダンスでは、AI医療機器の透明性確保が最重要課題として位置づけられています。
開発企業は、使用したデータセット、モデルアーキテクチャ、学習プロセス、性能評価結果を詳細に文書化する必要があります。
特に、AIの意思決定プロセスが医療従事者にとって理解可能な形で説明されることが求められており、単純な「ブラックボックス」システムは承認が困難になっています。
- 使用データセットの詳細な文書化
- モデルアーキテクチャの明確な説明
- 学習プロセスの透明性確保
- 性能評価結果の包括的な開示
モデルカードの作成と文書化の重要性

FDAが推奨するモデルカードには、AI医療機器の性能特性、限界、適用範囲が明確に記載される必要があります。
このモデルカードは、医療従事者がAI医療機器を適切に使用するための重要な情報源となります。また、患者の多様性を考慮した性能評価結果や、異なる医療環境での動作確認データも含める必要があります。
データセットの透明性と人口統計学的詳細の開示
AI医療機器の学習に使用されたデータセットの詳細な開示が義務化されています。人種、性別、年齢、地理的分布などの人口統計学的情報を明示し、データの代表性とバイアスの可能性を評価する必要があります。
また、データの収集方法、前処理手順、品質管理プロセスについても詳細な文書化が求められています。
信頼性評価フレームワークの実装方法
リスクベースアプローチによる評価基準
FDAはAI医療機器の評価において、リスクベースアプローチを採用しています。このアプローチでは、AI医療機器が患者に与える潜在的リスクの程度に応じて、要求される証拠のレベルが決定されます。
高リスク機器では、より厳格な臨床試験データが必要となり、低リスク機器では実世界データでの性能確認が認められる場合があります。
継続的なモニタリングと品質管理体制
AI医療機器の市販後監視では、継続的な性能モニタリングシステムの構築が必須となっています。
このシステムでは、実世界での使用データを収集・分析し、AI性能の変化や予期しない動作を早期に検出する必要があります。
また、定期的な性能評価レポートの提出と、必要に応じた迅速な是正措置の実施が求められています。
ReAlice株式会社 開発担当者FDAが求める文書化は、技術者にとってドキュメント主導の開発体制(Documentation-Driven Development)への意識改革を促すものです。継続監視システムの設計では、単なるログ取得にとどまらず動的なフィードバックループとして設計する視点が求められます。
FDA SaMD(Software as a Medical Device)とAI開発の実践的アプローチ
SaMDにおけるAI/ML技術の位置づけと分類
FDAのSaMD規制枠組みでは、AI/ML技術を搭載したソフトウェア医療機器が特別なカテゴリーとして位置づけられています。
従来のSaMDとは異なり、AI搭載SaMDは学習能力と適応性を持つため、静的なソフトウェアとは異なる評価アプローチが必要となります。FDAは、AI搭載SaMDを「Locked Algorithm」と「Adaptive Algorithm」に分類し、それぞれに適した規制要件を設定しています。
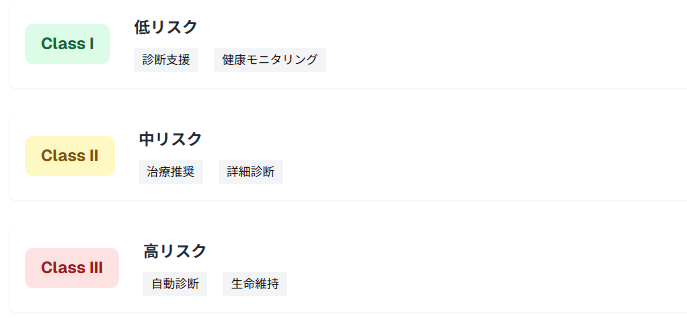
Class I、II、IIIによるリスク分類とAI適用範囲
AI搭載SaMDのリスク分類は、従来の医療機器分類に加えて、AIの自律性レベルと医療意思決定への影響度を考慮して決定されます。Class I(低リスク)では診断支援機能、Class II(中リスク)では治療推奨機能、Class III(高リスク)では自動診断・治療機能が主な適用範囲となっています。
この分類により、各リスクレベルに応じた適切な規制要件が適用されます。
510(k)申請とPMA承認の使い分け

AI搭載SaMDの市販前申請では、リスク分類と技術的新規性に基づいて510(k)申請またはPMA承認のいずれかが選択されます。
既存の医療機器と実質的に同等なAI機能を持つ場合は510(k)申請が可能ですが、革新的なAI技術や高リスク機能を持つ場合はPMA承認が必要となります。FDAは、AI技術の特性を考慮した新しい同等性評価基準を開発しており、従来の機械的同等性だけでなく、アルゴリズムの同等性も評価対象となっています。
SPSとACPによる変更管理プロトコル
SaMD Pre-Specificationsの設計要件
SaMD Pre-Specifications(SPS)は、AI医療機器の予定された変更を事前に定義し、承認を得るための重要な仕組みです。SPSでは、AIアルゴリズムの学習データ更新、性能改善、機能拡張などの変更範囲を明確に規定する必要があります。
また、各変更が患者安全性に与える影響を事前に評価し、適切なリスク軽減策を講じることが求められています。
Algorithm Change Protocolの実装戦略
Algorithm Change Protocol(ACP)は、AI医療機器のアルゴリズム変更を管理するための具体的な手順を定めたものです。
ACPの実装では、変更の種類、評価方法、承認プロセス、実装手順、監視方法が詳細に規定されます。
特に、機械学習モデルの再学習や性能調整において、患者安全性を確保しながら継続的改善を実現するためのバランスが重要となります。
ReAlice株式会社 開発担当者SPSやACPは、AIモデルの進化と規制遵守を両立させるための重要な設計要素であり、開発初期段階からの計画的導入が不可欠です。
CBER・CDER・CDRH・OCPの連携によるAI開発支援体制
各センターの役割分担と専門領域

FDAの4つの主要センターは、AI医療製品の包括的な規制体制を構築するため、密接に連携しています。
この連携体制により、医薬品から医療機器まで、AI技術を活用した幅広い医療製品に対する一貫した規制アプローチが実現されています。
各センターの専門性を活かしながら、横断的な標準化とベストプラクティスの共有が進められています。
- CDER:医薬品評価研究センター – 医薬品開発におけるAI活用支援
- CDRH:医療機器・放射線保健センター – AI搭載医療機器の規制
- CBER:生物学的製剤評価研究センター – バイオ医薬品のAI活用
- OCP:複合製品室 – AI技術を組み込んだ複合製品の規制
CDERによる医薬品開発におけるAI活用支援
CDER(医薬品評価研究センター)では、AI技術を活用した医薬品開発の効率化と品質向上を積極的に支援しています。
特に、臨床試験設計の最適化、患者層別化、安全性監視の自動化において、AIの活用が推進されています。FDAは2025年1月に「Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products」を発行し、医薬品開発におけるAI活用の具体的指針を示しました。
CDRHの医療機器・放射線機器評価体制
CDRH(医療機器・放射線保健センター)は、AI搭載医療機器の規制において最も先進的な取り組みを行っています。
2024年12月に発表されたPCCP(Predetermined Change Control Plan)ガイダンスでは、AI医療機器の継続的改善を可能にする革新的な規制枠組みが提示されました。このガイダンスにより、AI医療機器メーカーは事前に承認された変更計画に基づいて、迅速な性能改善を実現できるようになっています。
FDAは既に1,000件を超えるAI/ML医療機器を認可しており、市販後のソフトウェア更新ニーズが年々高まっている状況です。CDRHはこの急速な技術進歩に対応するため、従来の静的な承認プロセスから動的な変更管理システムへの転換を推進しています。
CBERの生物学的製剤評価とOCPの複合製品規制
CBER(生物学的製剤評価研究センター)では、AI技術を活用したバイオ医薬品開発の支援に注力しています。
抗体医薬品の設計最適化や細胞治療製品の品質管理において、AIの活用が期待されています。OCP(複合製品室)では、AI技術を組み込んだ複合製品(医薬品・医療機器・生物学的製剤の組み合わせ)の規制要件を整備しており、革新的な治療法の実現を支援しています。
部署間連携による統合的なAI規制アプローチ
横断的な標準化とベストプラクティスの共有
FDAの各センター間では、AI規制に関する知識とベストプラクティスの共有が活発に行われています。月次のAI規制会議では、各センターの経験と課題が共有され、統一的な規制アプローチの開発が進められています。
また、国際的な規制調和の観点から、EMA(欧州医薬品庁)やPMDA(医薬品医療機器総合機構)との連携も強化されています。
ReAlice株式会社 開発担当者FDAの各センターが役割を分担しつつ、AI製品に対する一貫した規制方針を築いている点は実務者にとって大きな安心材料となります。特にPCCPの導入やSPS/ACPといった柔軟な変更管理手法は、継続的に進化するAI技術に即した体制づくりといえます。
AI活用による医療製品開発の業務効率化メリット
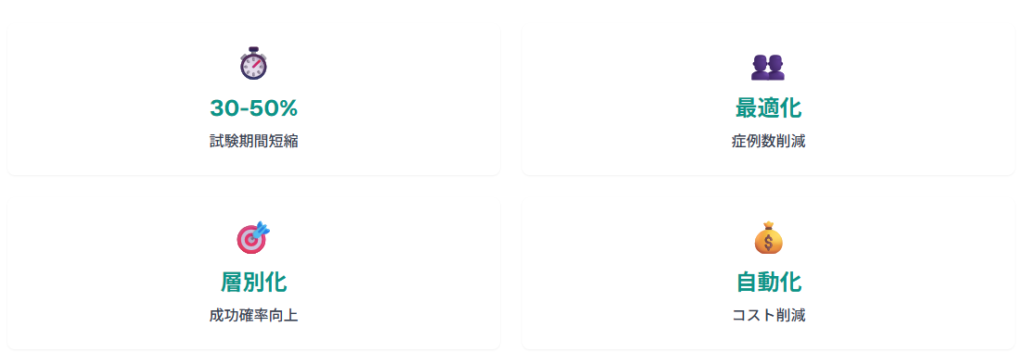
臨床試験設計の最適化と効率向上
AI技術の活用により、臨床試験の設計と実施効率が大幅に改善されています。
患者層別化、エンドポイント選択、試験期間短縮などの領域で、AIが重要な役割を果たしています。FDAのISAND Pilot Programでは、AI支援による臨床試験の効率化事例が多数報告されており、従来の試験設計と比較して30-50%の期間短縮が実現されています。
AIアルゴリズムを活用して、患者の遺伝的背景、病歴、バイオマーカーを分析し、治療効果が期待できる患者群を特定します。これにより、臨床試験の成功確率が向上し、必要な症例数を削減できます。
機械学習を用いて過去の臨床試験データを分析し、最も適切な主要エンドポイントと副次エンドポイントを選択します。これにより、試験の統計的検出力を向上させ、規制当局への説得力を高めます。
AI予測モデルを活用して、患者の治療反応を早期に予測し、中間解析の精度を向上させます。これにより、有効性が確認された時点で試験を早期終了し、開発期間を大幅に短縮できます。
デジタルヘルステクノロジー(DHT)の活用事例
デジタルヘルステクノロジーとAIの組み合わせにより、リモート患者モニタリングと自動データ収集が可能になっています。ウェアラブルデバイスから収集されるリアルタイムデータをAIが分析し、患者の状態変化を早期に検出することで、臨床試験の安全性と効率性が向上しています。
FDAは、DHT活用ガイダンスにおいて、AI分析結果の信頼性確保のための要件を明確化しています。
リアルワールドデータ(RWD)分析の実装
リアルワールドデータの活用において、AI技術は膨大なデータセットから有意義な洞察を抽出する重要な役割を担っています。
電子健康記録、保険請求データ、患者報告アウトカムなどの多様なデータソースを統合分析することで、医療製品の実世界での有効性と安全性を評価できます。FDAのSentinelシステムでは、AI技術を活用した大規模な安全性監視が実施されており、1億人以上のデータを基に効率的な有害事象分析を行っています。
薬事申請プロセスの効率化と品質向上
AI支援による申請資料作成の自動化
大手製薬企業では、AI技術を活用した申請資料の自動生成システムの導入が進んでいます。製薬業界では、規制申請プロセスの効率化と品質向上を目的として、AI技術の導入が積極的に検討されています。
このシステムでは、構造化されたデータ管理と自然言語生成技術を組み合わせることで、高品質な申請資料の迅速な作成を実現しています。
- 作成期間の効率化:AI技術による申請資料作成プロセスの改善
- 品質の向上:構造化データ管理による一貫性確保
- コスト削減:人的リソースの効率的活用
- 市場投入の加速:新薬の早期上市による競争優位性
規制当局との連携強化による承認期間短縮
AI技術を活用した申請資料の品質向上により、規制当局とのコミュニケーションが効率化されています。
FDAでは、AI支援による申請資料の事前審査システムを試験導入しており、申請の完全性チェックと初期評価の自動化を進めています。これにより、審査期間の短縮と審査品質の向上が同時に実現されています。
ReAlice株式会社 開発担当者臨床試験設計から申請資料作成まで、AIの導入が医薬品開発プロセスの根幹を効率化しています。
特に患者層別化やエンドポイント選定は、AIの強みが発揮される領域であり実世界データやDHTとの組み合わせにより信頼性も高まっています。
FDA AIファーマコビジランスと市販後安全性監視
AI技術を活用した有害事象検出システム
FDAは、AI技術を活用した革新的なファーマコビジランスシステムの構築を進めています。従来の手動による有害事象報告の処理では限界があったため、AI技術による自動化と効率化が急務となっていました。
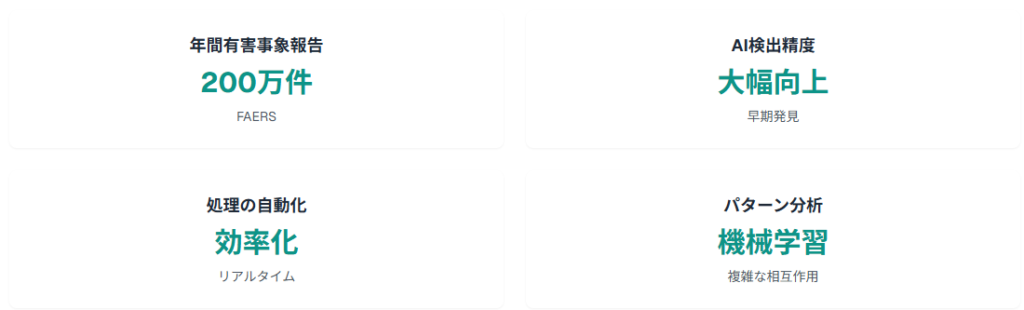
現在、FDAは年間約200万件の有害事象報告をFAERS(FDA Adverse Event Reporting System)で受け取っており、この膨大なデータの処理にAI技術が不可欠となっています。
- 年間200万件を超える有害事象報告の効率的処理
- 非構造化データからの重要情報抽出
- 稀な有害事象の早期検出
- 複雑な薬物相互作用パターンの特定
自動化された安全性シグナル検出の仕組み
FDAが開発したAI支援安全性監視システムでは、機械学習アルゴリズムが大量の有害事象報告から異常なパターンを自動検出します。
このシステムは、従来の統計的手法では発見困難だった稀な有害事象や複雑な薬物相互作用を早期に特定することが可能です。自然言語処理技術により、非構造化テキストデータからも重要な安全性情報を抽出し、包括的な安全性評価を実現しています。
機械学習による副作用パターン分析
AI技術を活用した副作用パターン分析では、患者の年齢、性別、併用薬、基礎疾患などの多様な要因を同時に考慮した高度な分析が可能となっています。
FDAのガイダンスによると、AI分析により従来の手法では見逃されていた安全性シグナルの検出精度が大幅に向上しており、患者安全の確保に重要な貢献をしています。
市販後監視におけるAI活用の実践例
リアルタイムデータ収集と分析体制
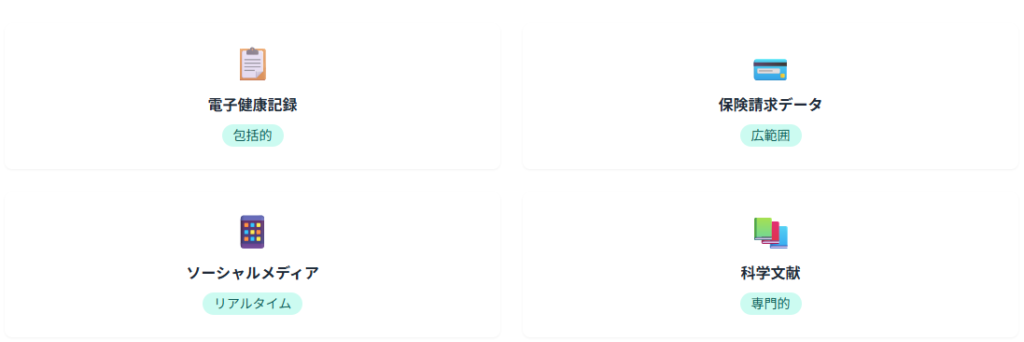
FDAは、リアルタイムでの安全性監視を実現するため、AI技術を活用したデータ収集・分析システムを構築しています。
このシステムでは、電子健康記録、保険請求データ、ソーシャルメディア、科学文献などの多様なデータソースから安全性情報を自動収集し、リアルタイムで分析を行います。
特に、AI医療機器の市販後監視では、MAUDE(Manufacturer and User Facility Device Experience)データベースの情報不足や分類の不備が課題として指摘されており、新たな監視体制の必要性が提起されています。
予測的安全性評価システムの構築
AI技術を活用した予測的安全性評価では、過去の安全性データと現在の使用パターンを分析し、将来の安全性リスクを予測します。このアプローチにより、重篤な有害事象が発生する前に予防的措置を講じることが可能となり、患者安全の向上と医療コストの削減を同時に実現しています。
FDAは、Total Product Lifecycle(TPLC)アプローチを採用し、市販前の開発から市販後の性能まで一連のパッケージとしてソフトウェア製品の評価とモニタリングを行っています。
ReAlice株式会社 開発担当者FAERSやMAUDEといった大規模データソースの処理には、AIのスケーラビリティとパターン認識力が不可欠です。自然言語処理と機械学習の併用により、非構造化報告や稀少事象からも安全性シグナルを高精度に抽出できるようになっています。
AI医療製品開発における具体的な活用事例と導入方法
成功事例から学ぶAI実装のベストプラクティス
AI医療製品開発の成功事例を分析すると、段階的な導入アプローチと継続的な改善プロセスが成功の鍵となっています。
特に、規制要件の早期理解と適切なリスク管理体制の構築が重要な成功要因として挙げられます。FDAは既に1,000件を超えるAI/ML医療機器を認可しており、市販後のソフトウェア更新ニーズが年々高まっている状況です。
大手製薬企業のAI活用戦略
Amgen社では、AI技術を活用した申請資料作成の自動化により、臨床試験プロセスの効率化を進めています。同社のAI戦略では、構造化コンテンツ管理システムと自然言語生成技術を組み合わせることで、申請資料作成プロセスの最適化を実現しています。
この取り組みにより、新薬の開発期間短縮への貢献が期待されています。
臨床試験データを構造化形式で管理し、AI処理に適した形でデータベースを設計。これにより、自動化処理の精度と効率性を大幅に向上させました。
高度な自然言語生成AIを導入し、規制要件に準拠した申請資料の自動作成を実現。人的リソースを戦略的業務に集中させることが可能になりました。
小規模なパイロットプロジェクトから開始し、成功事例を積み重ねながら全社的な展開を実現。リスクを最小化しながら効果を最大化する戦略を採用しました。
医療機器メーカーのSaMD開発事例
AI搭載医療機器の開発では、FDAのPCCP(Predetermined Change Control Plan)ガイダンスを活用した継続的改善アプローチが注目されています。患者モニタリングソフトウェアを開発した企業では、PCCPを活用してAIモデルの再学習による偽陽性率の削減を実現しました。
従来であれば新たな510(k)申請が必要だった変更を、事前承認された変更計画に基づいて迅速に実装することで、市場競争力を維持しています。
中小企業でも実現可能なAI導入アプローチ
段階的な導入計画の立案方法
中小企業におけるAI導入では、限られたリソースを効率的に活用するため、段階的なアプローチが重要です。
まず、既存の業務プロセスの中でAI化による効果が最も期待できる領域を特定し、小規模なパイロットプロジェクトから開始することが推奨されます。
FDAのEmerging Drug Safety Technology Program(EDSP)では、中小企業向けの相談窓口を設置し、AI導入に関する規制相談を受け付けています。
コスト効率的なAI開発リソースの活用
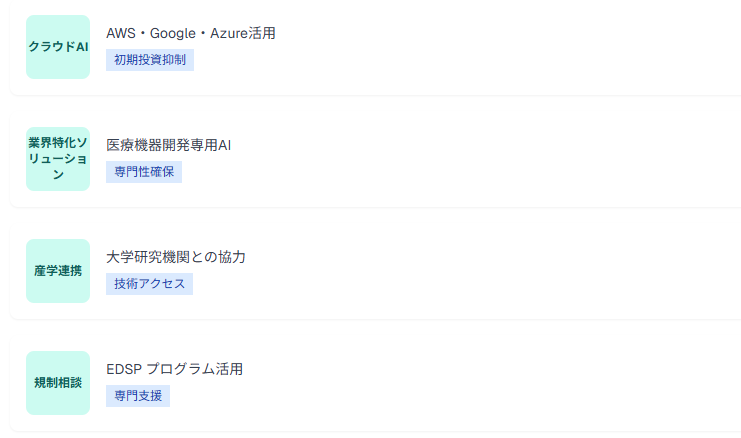
中小企業では、自社でのAI開発に加えて、外部リソースの活用が重要な選択肢となります。クラウドベースのAIプラットフォームサービスや、業界特化型のAIソリューションを活用することで、初期投資を抑えながらAI技術を導入できます。
また、大学や研究機関との連携により、最新のAI技術へのアクセスと専門知識の獲得が可能となります。
- クラウドAIサービス:AWS、Google Cloud、Microsoft Azureの医療特化AIツール
- 業界特化ソリューション:医療機器開発専用のAIプラットフォーム活用
- 産学連携:大学研究機関との共同研究による技術アクセス
- 規制相談活用:FDAのEDSPプログラムによる中小企業支援
ReAlice株式会社 開発担当者Amgenのようにデータ構造の設計から着手することで、AI処理の再現性と品質が確保されます。
また、PCCPやEDSPといったFDAの仕組みを活用すれば、中小企業でも計画的なAI展開が実現可能です。
FDA AI規制対応のための開発戦略と将来展望
2025年以降の規制動向予測と対応準備
FDAのAI規制は2025年を境に大きな転換点を迎えており、今後さらに詳細化・具体化が進むと予想されます。
現在パブリックコメント期間中のガイダンス草案が正式版として発行されることで、AI医療製品開発の規制要件が確定し、業界全体の開発戦略に大きな影響を与えることが予想されます。
特に、2025年1月に発表された「Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management」ドラフトガイダンスでは、PCCP(Predetermined Change Control Plan)の具体的な実装方法が明確化されています。
新たなガイダンス策定スケジュールと影響範囲
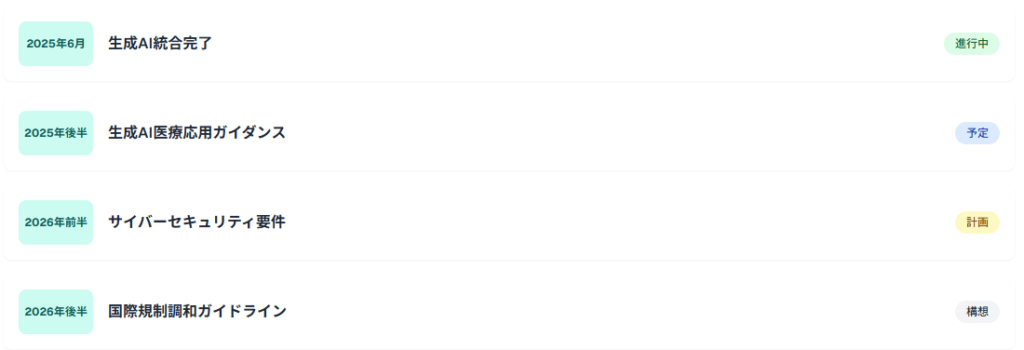
FDAは2025年後半から2026年にかけて、AI医療製品に関する追加ガイダンスの発行を予定しています。特に、生成AI技術の医療応用、AI医療機器のサイバーセキュリティ要件、国際的な規制調和に関するガイダンスが重点項目として挙げられています。
これらのガイダンスは、次世代AI技術の医療応用を促進する一方で、患者安全の確保を最優先とした規制枠組みを提供することが期待されています。
- 2025年6月:生成AI内部利用の全センター統合完了
- 2025年後半:生成AI医療応用ガイダンス発行予定
- 2026年前半:サイバーセキュリティ要件ガイダンス
- 2026年後半:国際規制調和ガイドライン策定
国際的な規制調和への取り組み
FDAは、EMA、PMDA、Health Canadaなどの国際的な規制当局と連携し、AI医療製品に関する規制の調和を進めています。
国際的な規制調和の必要性は各国で認識されており、AI医療技術の急速な発展に対応するため、規制アプローチの調和に向けた議論が継続されています。
欧州では、MDR/IVDRとAI法という二つの重要な規制枠組みが存在し、AI医療機器に関する規制の明確化が求められています。
AI技術の進化に対応した長期的開発戦略

次世代AI技術(生成AI等)への規制対応
生成AI技術の医療応用は、従来のAI技術とは異なる新たな課題と機会をもたらしています。
FDAは、生成AIの創造性と予測不可能性を考慮した新しい評価手法の開発を進めています。医療従事者向けの診断支援や治療計画立案における生成AI活用が期待される一方で、出力の信頼性確保と責任の所在明確化が重要な課題となっています。
FDAは2025年1月に「Considerations for the Use of Artificial Intelligence to Support Regulatory Decision-Making for Drug and Biological Products」を発行し、AI技術の規制における活用指針を示しました。
FDAは、AI技術による規制業務の効率化と品質向上を目指しており、科学的レビュープロセスの改善に取り組んでいます。
持続可能なAI開発エコシステムの構築
長期的なAI医療製品開発の成功には、産学官連携による持続可能なエコシステムの構築が不可欠です。
FDAは、大学研究機関、医療機関、テクノロジー企業、規制当局が連携する新しい協力モデルの構築を支援しており、革新的なAI技術の安全で効率的な医療応用を促進しています。
特に、新興医薬品安全技術会議(EDSTM)プログラムを通じて、医薬品安全性監視における新しい技術とその潜在的な応用について議論するプラットフォームを提供しています。
- 産学連携の強化:大学研究機関との共同研究による技術革新
- 国際協力の推進:グローバルな規制調和による開発効率向上
- 継続的学習システム:リアルワールドデータを活用した性能改善
- 透明性の確保:AIモデルの説明可能性と信頼性向上
ReAlice株式会社 開発担当者2025年以降のFDAのAI規制強化は、医療AI開発における転換期となり、特に生成AIやサイバーセキュリティ領域での新指針は実装戦略に直結します。
FDA AIに関してよくある質問
FDAとは何で、AI規制にどのような役割を果たしていますか?
FDA(米国食品医薬品局)は、米国保健福祉省の一部門として、食品、医薬品、医療機器、生物学的製剤などの安全性と有効性を確保する連邦機関です。
AI技術の医療応用においては、革新的技術の促進と患者安全の確保のバランスを取りながら、包括的な規制枠組みを構築しています。FDAは2025年に入ってから、AI医療製品に特化した複数のガイダンス草案を発表し、業界の技術革新を支援しながら適切な規制監督を実施しています。
現在、FDAは1,000件を超えるAI/ML医療機器を既に認可しており、市販後のソフトウェア更新ニーズが年々高まっている状況に対応しています。
CDERとはFDAの何で、AI医薬品開発にどう関わりますか?
CDER(Center for Drug Evaluation and Research:医薬品評価研究センター)は、FDAの主要部門の一つで、医薬品の安全性と有効性の評価を担当しています。
AI医薬品開発においては、臨床試験設計の最適化、患者層別化、安全性監視の自動化を支援するAI技術の活用を推進しています。CDERは「Good Machine Learning Practices」ガイドラインを通じて、医薬品開発におけるAI活用の標準的手法を確立し、業界の技術革新を支援しています。
また、2025年1月に発表されたドラフトガイダンス「医薬品および生物学的製剤の規制判断を支援するためのAI活用に関する検討事項」では、リスクベースの信頼性評価フレームワークを提示しています。
FDA SaMDとは何で、AI医療機器開発にどのような影響がありますか?
SaMD(Software as a Medical Device)は、ハードウェア医療機器に組み込まれることなく、単独で医療目的に使用されるソフトウェアを指します。
AI搭載SaMDは、従来の静的ソフトウェアとは異なり、学習能力と適応性を持つため、FDAは特別な規制要件を設定しています。PCCPガイダンスにより、AI医療機器の継続的改善が可能となり、事前承認された変更計画に基づく迅速な性能向上が実現されています。
AI関連申請は2016年以降急激に増加しており、放射線画像診断、ECG解析、内視鏡支援などの分野でAI技術の活用が活発に進んでいます。
FDA AI transparencyの要求事項と実装方法は?
FDAのAI透明性要求では、AIモデルの開発プロセス、使用データ、性能特性、限界を詳細に文書化することが必要です。
具体的には、モデルカードの作成、データセットの人口統計学的詳細の開示、アルゴリズムの意思決定プロセスの説明可能性確保が求められています。実装では、開発段階からの継続的な文書化、多様な患者集団での性能評価、医療従事者向けの理解しやすい説明資料の準備が重要となります。
FDAが内部でAIツール(例:cderGPT)の利用を進めるにつれて、スポンサーが提出するAIモデルに対する審査も、より洗練され詳細なものになることが予想されます。
AI医療製品の承認プロセスと必要な準備は?
AI医療製品の承認プロセスは、リスク分類に応じて510(k)申請またはPMA承認のいずれかが選択されます。
準備段階では、適切なリスク評価、臨床データの収集、透明性要件の充足、サイバーセキュリティ対策の実装が必要です。特に、AI特有の継続的学習機能を持つ製品では、SPSとACPの策定により、市販後の変更管理計画を事前に承認取得することが推奨されています。
また、FDAとの早期連携が重要であり、特にリスクの高い申請については、新興医薬品安全技術会議(EDSTM)プログラムを活用した事前相談が効果的です。
市販後監視では、MAUDEデータベースの情報不足や分類の不備が課題として指摘されており、新たな監視体制の構築が進められています。
