
FDA開発AI規制ガイド|医療機器承認から申請プロセスまで徹底解説

「FDA開発AIって何?」「AI医療機器の承認にはどんな手続きが必要なの?」「日本企業が米国市場に参入するにはどうすればいい?」そう思う方も多いのではないでしょうか。
実は、FDAのAI医療機器承認は2024年だけで107件を突破し、総承認数は950を超えるなど急速に拡大しています。しかし、従来の医療機器とは異なるAI特有の規制要件や申請プロセスを理解せずに参入を試みると、承認取得に想定以上の時間とコストがかかってしまう可能性があります。
本記事では、FDA開発AI規制の基本概念から具体的な申請プロセス、サイバーセキュリティ対策、そして日本企業の米国市場参入戦略まで、AI医療機器の開発・承認に必要な情報を包括的に解説します。AIメディカルサービス社のブレークスルーデバイス指定などの実際の承認事例も交えながら、実践的なノウハウをお伝えしていきます。
- FDA開発AIの基本概念と規制フレームワーク
- AI医療機器の承認プロセス
- 最新のFDAガイダンスとサイバーセキュリティ要件
- 日本企業の米国市場参入戦略
- AI開発による業務効率化のメリットと将来性

FDA開発AIの基本概念と規制フレームワーク
FDAが定義するAI搭載医療機器とは
FDAは、機械学習やディープラーニングアルゴリズムを使用して医療診断、治療計画、患者モニタリングを支援するソフトウェアをAI搭載医療機器として分類しています。
2025年1月6日に発表された最新ドラフトガイダンス「Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations」では、これらのデバイスの定義がより明確化されました。
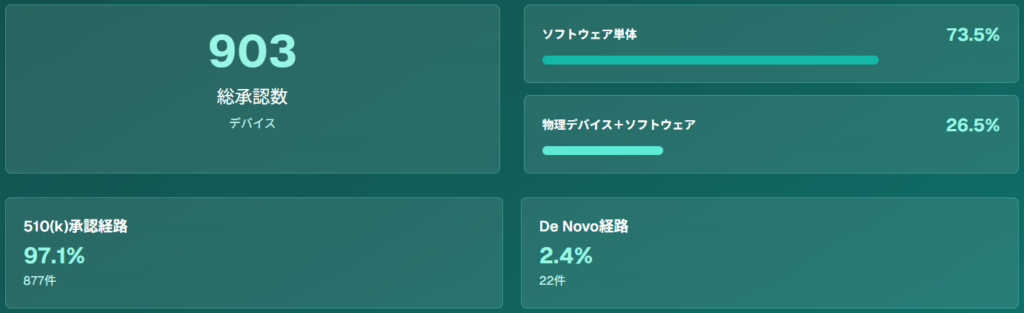
特に注目すべきは、これらのデバイスの97.1%が510(k)承認経路を通じて承認されており、従来の医療機器承認プロセスが効果的に機能していることを示しています。
AI/ML医療機器の分類とリスクベースアプローチ
FDAは、AI/ML医療機器をリスクレベルに応じて分類し、それぞれに適切な規制要件を適用するリスクベースアプローチを採用しています。
現在承認されている医療機器の分野別内訳を見ると、放射線科が76.6%と圧倒的多数を占め、心血管系が10.1%、神経科が3.2%と続いています。
- 画像診断支援AIが最も実用化が進んでいる分野
- 新たなサイバーセキュリティ要件を導入
- 1億8500万件の医療記録侵害を受けて規制強化
FDAは2024年に新たなサイバーセキュリティ要件を導入し、昨年だけで1億8500万件の医療記録が侵害された現状を受けて、AI搭載デバイスに対してより厳格なセキュリティ対策を求めています。
Total Product Lifecycle(TPLC)アプローチの重要性
FDAのTotal Product Lifecycle(TPLC)アプローチは、AI医療機器の開発から市販後まで一貫した品質管理を要求する包括的なフレームワークです。このアプローチの核心は、事前変更管理計画(Predetermined Change Control Plan: PCCP)の策定にあります。
PCCPにより、製造業者はAIモデルの更新を事前に計画し、重大な性能変更でない限り、長期間の再承認プロセスを経ずにリアルワールドデータに基づく改善を実施できます。
Hologic社のGenius™ Cervical AI algorithmは、このアプローチを活用した成功例として、従来の顕微鏡検査と比較して偽陰性を28%削減し、遠隔での症例レビューを可能にしました。
 ReAlice株式会社 開発担当者
ReAlice株式会社 開発担当者セキュリティ要件の強化はAI搭載機器の遠隔運用における脆弱性リスクに対する実務的な対策です。臨床研究でのバイアス低減に向け、サブグループ解析の標準化も今後の課題です。
FDA開発AI承認事例と最新動向

放射線科領域でのAI医療機器承認事例
放射線科領域では、Hologic社のGenius™ Digital Diagnostics Systemが2024年2月1日にFDA承認を取得し、子宮頸がんスクリーニングの精度向上に大きく貢献しています。このシステムは、従来のパップテスト用ガラススライドをデジタル化し、AIアルゴリズムを適用することで、高悪性度扁平上皮内病変およびより重篤な病変の検出において偽陰性を28%削減しました。

Canon、Siemens、GE、Philipsといった老舗医療機器メーカーが医療AI分野で活動しており、GEは3年連続でFDAのAI対応医療機器承認数において首位を維持しています。
これらの企業のうち、Siemens(1847年創業)、GE(1892年創業)、Philips(1891年創業)は19世紀創業の歴史ある企業ですが、Canon(1933年創業)は20世紀の企業です。
心血管・神経科領域での承認実績
心血管領域では、Powerful Medical社のPMcardio STEMI AI ECGモデルが2025年3月にFDAブレークスルーデバイス指定を受けました。このシステムは、ST上昇型心筋梗塞(STEMI)とSTEMI等価病変をECGで検出し、特に専門医による即座の評価が困難な地方医療機関での診断精度向上を目指しています。
- 地方医療センターでの適切な治療を受けられる患者:わずか17%
- Edwards Lifesciences社のEvoque三尖弁置換システムも承認
- TRISCEND II試験で三尖弁逆流の改善を実証
地方医療センターに搬送される患者のうち、適切な時間内にカテーテル室での治療を受けられるのはわずか17%という現状を改善する重要な技術として期待されています。
Edwards Lifesciences社のEvoque三尖弁置換システムも2024年に米国初の経カテーテル三尖弁デバイスとしてFDA承認を取得し、TRISCEND II試験で三尖弁逆流の改善と患者症状の有意な改善を実証しました。
2024年度FDA承認AI医療機器の統計データ
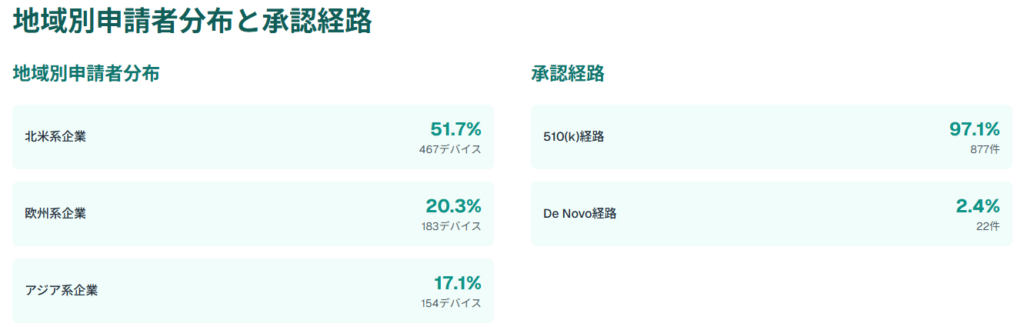
2024年8月31日時点の統計データによると、FDA承認AI医療機器の総数は903に達し、そのうち860が臨床使用可能、43がリコール対象となっています。地域別の申請者分布では、北米系企業が51.7%(467デバイス)と過半数を占め、欧州系が20.3%(183デバイス)、アジア系が17.1%(154デバイス)となっています。

臨床性能研究が実施されたデバイスは55.9%(505デバイス)で、そのうち38.2%が後ろ向き研究、8.1%が前向き非無作為化研究、2.4%が無作為化研究でした。
注目すべきは、性別サブグループの情報が利用可能なのは28.7%、年齢関連サブグループの情報が利用可能なのは23.2%に留まっており、多様性を考慮した臨床研究の必要性が浮き彫りになっています。
ReAlice株式会社 開発担当者AIの臨床応用が進む中、放射線・心血管領域では特に画像診断や信号解析に強いAIモデルが成果を上げています。
AI開発におけるFDAガイダンスの詳細解説
FDAのAI医療機器に関するガイダンスは、急速に進化するAI技術に対応するため継続的に更新されており、開発者にとって実践的な指針を提供しています。
最新のガイダンスでは、従来の医療機器開発とは異なるAI特有の課題への対応策が詳細に示されています。
2025年1月発表の最新ドラフトガイダンス
2025年1月7日にFDAが発表した「Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations」は、AI搭載医療機器の開発から市販後管理まで包括的に扱うドラフトガイダンスです。同月14日には、このガイダンスに関する説明会(Webinar)が開催されました。
このガイダンスは、AI医療機器が継続的に学習・適応する特性を考慮し、従来の静的な医療機器とは異なるライフサイクル管理アプローチを提示しています。
- 透明性とバイアス制御の中心原則
- リスクベースアプローチの採用
- データ管理、文書化、ラベリング、サイバーセキュリティプロセス
- 開発から市販後管理まで一貫した品質保証
特に重要なのは、Total Product Lifecycle(TPLC)フレームワークの強調で、AIシステムが時間の経過とともに安全で効果的であり続けることを保証します。透明性とバイアス制御の中心原則を重視し、リスクベースアプローチの重要性を強調しています。
事前変更管理計画(PCCP)の作成方法
事前変更管理計画(PCCP)は、AI医療機器の継続的改善を可能にするメカニズムです。
2024年12月にFDAがPCCPに関するガイダンスを最終化し、製造業者がリアルワールドデータに基づくAIモデルの更新を、デバイス性能に重大な変更をもたらさない限り、長期間の再承認プロセスを経ずに実施できるようになりました。
どのような更新が事前承認の範囲内かを具体的に示す必要があります。アルゴリズムの微調整、データセットの拡張、性能指標の改善など、具体的な変更内容を明確に定義します。
感度、特異度、AUC(Area Under the Curve)などの定量的指標に加え、臨床的有用性を評価する定性的指標も含める必要があります。
- 定量的指標:感度、特異度、AUC値
- 定性的指標:臨床的有用性、患者満足度
- リアルタイム性能監視システム
Boston Scientific社のFARAPULSE PFA Systemは、ADVENT研究と実世界データに基づく承認を受け、より短い手術時間と重篤な副作用の回避を実現した成功例として注目されています。
Good Machine Learning Practice(GMLP)の実践

Good Machine Learning Practice(GMLP)は、AI医療機器開発における品質保証の基盤となる実践指針です。GLMPの核心は、高品質で代表性のあるデータセットの構築にあり、バイアスを排除し多様性を確保することが重要です。
- Prenosis社のSepsis ImmunoScore:初のAI敗血症診断ツール
- 2024年4月にFDAのDe Novo承認を受けた
- 精密医療プラットフォームImmunixを活用
- 急性期医療における個別化医療の実現を目指す
GMLP実践では、アルゴリズムの説明可能性も重要で、医療従事者がAIの判断根拠を理解できるよう、ブラックボックス化を避ける設計が求められます。継続的な性能監視により、アルゴリズムドリフトやデータポイズニングなどの問題を早期発見し、適切な対策を講じることも必須要件となっています。
ReAlice株式会社 開発担当者FDAの最新ガイダンスは、AIシステムの変化に対応した動的な品質管理を可能にする点が非常に実践的です。PCCPのようなメカニズムにより再承認の負担を軽減しつつ、安全性を確保する設計が進化しています。
FDA開発AI申請プロセスの実務対応
FDAのAI医療機器申請プロセスは、従来の医療機器とは異なる独特の要件と手順を持っており、成功のためには戦略的なアプローチが必要です。
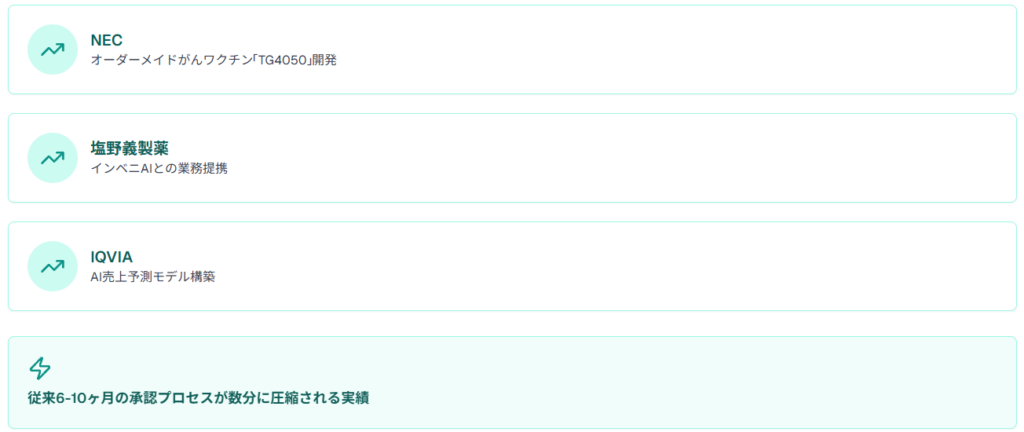
2025年1月7日にFDAが発表したAI医療機器ガイダンスは、AI搭載医療機器の開発から市販後管理まで包括的な指針を提供しています。この動きにより、従来6-10ヶ月かかっていた新薬承認プロセスにおいて、AI技術により3日間の審査作業が数分に圧縮される実績が報告されています。
510(k)申請でのAI医療機器承認要件
510(k)申請経路は、AI医療機器承認の主流となっており、2024年8月時点で903のAI搭載医療機器のうち877件(97.1%)がこの経路で承認されています。510(k)申請では、既存の合法的に販売されている医療機器(predicate device)との「実質的同等性」を証明する必要があります。
- アルゴリズムの性能指標の比較
- 学習データセットの特性評価
- 臨床的有用性の証明
AI医療機器の場合、アルゴリズムの性能指標、学習データセットの特性、臨床的有用性の比較が重要な評価ポイントとなります。Powerful Medical社のPMcardio STEMI AI ECGモデルは、2025年3月にFDAブレークスルーデバイス指定を受け、地方医療センターでのST上昇型心筋梗塞検出において、専門医による即座の評価が困難な状況での診断精度向上を実現しました。
現在、地方医療センターに搬送される患者のうち、適切な時間内にカテーテル室での治療を受けられるのはわずか17%という課題を解決する技術として期待されています。
De Novo分類とPMA申請の使い分け
De Novo分類は、既存のpredicate deviceが存在しない新しいAI医療機器に適用される経路です。Prenosis社のSepsis ImmunoScoreは、2024年4月3日にFDAのDe Novo承認を受けた初のAI敗血症診断ツールとして、精密医療プラットフォームImmunixを活用した個別化医療の実現を目指しています。
- De Novo分類:新規デバイス向け(2.4%)
- PMA申請:最高リスククラスIII向け
- Edwards Lifesciences社のEvoque三尖弁置換システムがPMA承認
- TRISCEND II試験で有効性を実証
一方、PMA申請は最もリスクの高いクラスIII医療機器に適用され、広範囲な臨床試験データによる安全性と有効性の証明が必要です。
Edwards Lifesciences社のEvoque三尖弁置換システムは、2024年に米国初の経カテーテル三尖弁デバイスとしてPMA承認を取得し、TRISCEND II試験で三尖弁逆流の改善と患者症状の有意な改善を実証しました。
臨床試験デザインとリアルワールドデータ活用
AI医療機器の臨床試験では、従来の医療機器とは異なる考慮事項があります。2024年8月時点の統計によると、臨床性能研究が実施されたAI医療機器は55.9%(505デバイス)で、そのうち38.2%が後ろ向き研究、8.1%が前向き非無作為化研究、2.4%が無作為化研究でした。
- 性別サブグループ情報:28.7%のみ利用可能
- 年齢関連サブグループ情報:23.2%のみ利用可能
- 多様性を考慮した臨床研究の必要性が浮き彫り
注目すべきは、性別サブグループの情報が利用可能なのは28.7%、年齢関連サブグループの情報が利用可能なのは23.2%に留まっており、多様性を考慮した臨床研究の必要性が浮き彫りになっています。
人種・民族情報が利用可能なのはわずか15.8%であり、AI医療機器の一般化可能性を確保するためには、より包括的な多様性データの収集が求められています。
ReAlice株式会社 開発担当者AI医療機器のFDA申請では、従来型の静的デバイスとは異なり、動的に学習・進化する特性を持つアルゴリズムの信頼性評価が鍵になります。510(k)の実質的同等性だけでなく、学習データの質とバイアス管理が重要です。
AI開発におけるサイバーセキュリティ対策

AI医療機器のサイバーセキュリティは、患者安全と医療システムの信頼性を確保するため、FDAが最重要視する分野の一つです。2024年だけで1億8500万件の医療記録が侵害された現状を受けて、FDAは新たなサイバーセキュリティ要件を導入しました。
FDAが求めるAI医療機器のセキュリティ要件
FDAの新しいサイバーセキュリティ要件では、AI医療機器製造業者に対して包括的なセキュリティ計画の策定を義務付けています。これには、脅威モデリング、脆弱性評価、インシデント対応計画、継続的監視システムの実装が含まれます。
特にAI医療機器では、学習データの完全性保護とモデルの改ざん防止が重要な要素となっています。
GMLP(Good Machine Learning Practice)の第2原則として、適切なソフトウェア工学及びセキュリティ対策の実施が求められています。これには、データ管理、文書化、ラベリング、サイバーセキュリティプロセスが含まれます。
GMLP第10原則では、上市されたモデルの性能をモニタリングし、再学習のリスクを管理することが求められています。これにより、新たな脅威に対する迅速な対応が可能になります。
データポイズニングと敵対的攻撃への対策
AI医療機器特有のセキュリティ脅威として、データポイズニング攻撃と敵対的攻撃への対策が必要です。
データポイズニング攻撃では、悪意のある者が学習データに意図的に偽の情報を混入させ、AIモデルの判断を誤らせる可能性があります。敵対的攻撃では、人間には識別困難な微細な変更を医療画像に加えることで、AIの診断結果を操作する危険性があります。
- AIを駆使したより巧妙なフィッシング攻撃
- リアルタイムでマルウェアを進化させる攻撃
- IoT機器の数が320億台に達する予測
- 量子コンピューターによる暗号技術への影響
効果的な対策には、データの出所確認、異常検知システムの実装、モデルの堅牢性テストが含まれます。
Hologic社のGenius™ Digital Diagnostics Systemでは、デジタル化されたパップテストスライドの処理において、データ完全性チェックと異常パターン検出により、偽陰性を28%削減しながらセキュリティを確保しています。
継続的監視とペネトレーションテストの実施
AI医療機器のセキュリティは、一度設定すれば終わりではなく、継続的な監視と評価が必要です。
FDAは、製造業者に対して定期的なペネトレーションテストの実施と脆弱性評価の更新を求めています。これには、新たに発見された脅威への対応、セキュリティパッチの適用、インシデント対応計画の定期的な見直しが含まれます。
- 2025年1月7日発表の最新ガイダンスでTPLCアプローチを強調
- 透明性とバイアス制御の中心原則を重視
- リスクベースアプローチの重要性を強調
2025年1月7日にFDAが発表した最新ドラフトガイダンス「Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations」では、Total Product Lifecycle(TPLC)フレームワークの重要性が強調されており、AIシステムが時間の経過とともに安全で効果的であり続けることを保証する包括的なアプローチが示されています。
このような透明性とトレーサビリティの確保は、AI医療機器のセキュリティ対策においても重要な要素となっています。
ReAlice株式会社 開発担当者AI医療機器では、学習済みモデルの堅牢性だけでなく、リアルタイムな異常検知と脆弱性管理の仕組みが不可欠です。特に、データポイズニングや敵対的攻撃のようなAI特有のセキュリティリスクに対しては、データクレンジングや推論時の検証ロジックの強化が求められます。
日本企業のFDA開発AI市場参入戦略

日本企業がFDA開発AI市場に参入する際は、規制環境の違いと市場特性を深く理解した戦略的アプローチが必要です。
2023年4月、日本では医療機器基本要件基準が改定され、プログラムを使用した医療機器へのサイバーセキュリティ対策が必須となりました。この規制強化により、日本企業は国内でのAI医療機器開発経験を活かしながら、米国市場への展開を図る機会が拡大しています。
PMDAとFDAの規制要件の違い
PMDAとFDAの規制要件には、承認プロセスと技術的要求事項において重要な違いがあります。PMDAでは、2023年4月の改定により、AI医療機器に対してIEC 81001-5-1規格に基づくサイバーセキュリティ対策が義務化されました。
一方、FDAは2025年1月7日に発表した最新ガイダンス「Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations」で、事前変更管理計画(PCCP)を重視したライフサイクル管理アプローチを採用しています。
IEC 81001-5-1規格準拠のサイバーセキュリティ対策が必須となり、医療機器基本要件基準の改定(2023年4月)により段階的な規制強化アプローチを採用しています。
Total Product Lifecycle(TPLC)アプローチを採用し、事前変更管理計画(PCCP)を重視しています。また、リアルワールドデータの積極的活用を推進しています。
日本企業にとって特に注意すべきは、FDAがリアルワールドデータの活用をより積極的に推進している点で、Boston Scientific社のFARAPULSE PFA Systemのように実世界データに基づく承認事例が増加しています。また、FDAは2024年だけで1億8500万件の医療記録侵害を受けて、より厳格なサイバーセキュリティ要件を導入しており、日本企業は両国の規制要件を同時に満たす設計が求められます。
米国市場参入時の留意点と成功事例
米国市場参入において、日本企業は文化的・技術的な違いを理解した戦略が必要です。
特に重要なのは、米国の医療現場における実用性とコスト効率性の重視で、技術的優位性だけでは市場浸透が困難です。株式会社日経リサーチが2023年3月に発表した「医療情報システム導入状況」調査では、AIやロボットといった最先端のデジタル技術への期待が高まっていることが確認されています。
- AIを駆使したより巧妙なフィッシング攻撃
- リアルタイムでマルウェアを進化させる攻撃
- IoT機器の数が416億台に達する予測
- 量子コンピューターによる暗号技術への影響
- 地政学的リスクに起因するサイバー攻撃
また、2025年のセキュリティ予測では、攻撃者はAIを使ってリアルタイムでマルウェアを進化させ、検出されにくくする技術を開発しており、従来のセキュリティ対策では対応しきれないという危険が増しています。このような環境下で、日本企業は迅速な市場投入が可能になっています。
現地パートナーシップの活用方法
効果的な現地パートナーシップは、日本企業の米国市場参入成功の鍵となります。特に重要なのは、規制対応に精通した現地コンサルティング企業との連携で、FDAの複雑な申請プロセスを効率的に進めることができます。
- 規制対応専門コンサルティング企業との連携
- 現地医療機関との臨床試験パートナーシップ
- 現地法律事務所との知的財産権保護連携
- 技術移転とリスク最小化の戦略構築
Powerful Medical社のPMcardio STEMI AI ECGモデルが2025年3月にブレークスルーデバイス指定を受けた事例のように、現地の医療ニーズを深く理解したパートナーとの協力により、適切な申請戦略を構築できます。
また、現地の医療機関との臨床試験パートナーシップも重要で、Edwards Lifesciences社のEvoque三尖弁置換システムがTRISCEND II試験を通じて承認を取得した例のように、信頼性の高い臨床データの収集が可能になります。さらに、技術移転や知的財産権の保護において、現地法律事務所との連携により、リスクを最小化しながら市場参入を実現できます。
ReAlice株式会社 開発担当者現地パートナーとの連携は、単なる規制対応に留まらず、実臨床との接続性を高めるブリッジでもあります。
AI開発による業務効率化のメリットと将来性
AI開発による業務効率化は、医療機器業界全体に変化をもたらしており、その効果は開発プロセスから規制対応まで広範囲に及んでいます。
2025年1月開始予定のジェトロ「HealthTech Gateway “AI Medical in the US”」プログラムでは、Mayo Clinic Platformと連携し、日本のデジタルヘルススタートアップ16社の米国市場参入を支援しています。この変化により、製薬企業や医療機器メーカーは、より迅速な製品開発と市場投入が可能になっています。
医療機器開発プロセスの効率化効果
AI技術の導入により、医療機器開発の各段階で大幅な効率化が実現されています。設計段階では、AIによるシミュレーションと予測モデリングにより、従来の物理的プロトタイプ作成に要していた時間とコストを大幅に削減できます。
日本電気株式会社(NEC)とTransgene社が共同開発したがんワクチン「TG4050」では、患者の正常な細胞とがん細胞とを比較し、がん細胞だけに見られる異常タンパク質「ネオアンチゲン」をAIによって予測するオーダーメイド型治療法を実現しました。
- NEC:AIを活用したオーダーメイドがんワクチン「TG4050」開発
- 塩野義製薬:インベニAIとの業務提携で精神・神経系疾患治療薬候補探索
- IQVIA:ブレインパッドとのAI売上予測モデル構築
- Mayo Clinic Platform:300万人以上の非識別化医療データ活用
また、塩野義製薬株式会社は、AIを使った創薬技術を持つ米国の企業インベニAIと業務提携し、精神・神経系の疾患の治療薬候補を探索しています。インベニAIの「AlphaMeld」プラットフォームは、過去10年以上にわたり培われたデータセットをもとにして構築された機械学習アルゴリズムで、特定の疾患に関連する標的分子の選定だけでなく、関連する複数の標的やその標的に対しての既存薬の提唱も可能です。
規制対応業務でのAI活用可能性
規制対応業務におけるAI活用は、製薬・医療機器業界の生産性向上に大きく貢献しています。米国では、FDA(食品医薬品局)によるAI医療機器の承認件数が累計1,000件を突破し、スマートウォッチを活用した心房細動の早期検出アルゴリズムや、眼底画像を用いた糖尿病網膜症検出AIなどが実用化されています。
- 米国:FDA承認AI医療機器が累計1,000件突破
- 欧州:オリンパスのクラウド型内視鏡AIシステムがCEマーク取得
- 日本:胃癌の深達度診断支援内視鏡AIがPMDA承認
- DASH for SaMDプログラムで医療ソフトウェア実用化促進
企業側でも、IQVIAソリューションジャパン株式会社と株式会社ブレインパッドが協力し、AIによる売上予測モデルの構築を導入して製薬会社の意思決定を支援しています。
将来の売上予測値は、クライアント企業からのニーズが高いことからサービス化を実施し、機械学習モデルを構築して算出することで効率的な対応が可能になっています。
次世代AI技術とFDA規制の展望
次世代AI技術の発展に伴い、FDA規制も継続的に進化しており、2025年以降の医療AI分野では更なる進歩が期待されています。
米国のXaira TherapeuticsはシリーズAで10億ドルの資金調達を成功させるなど、業界史上最大級の調達事例も登場しています。また、大手医療機器企業によるM&Aも積極化しており、米国のQuest DiagnosticsがPathAIを買収し、AIを診断業務に積極的に活用する方針を示しています。

今後の医療AIでは、基盤モデルの医療応用が大きな注目を集めるでしょう。複数の異なる医療データを学習し、多面的な診断支援を可能にする技術で、診断や創薬の精度向上が期待されています。
しかし、大規模モデルにはブラックボックス化やバイアスのリスクも伴うため、「説明可能なAI(XAI)」や公平性を担保する技術開発が不可欠です。また、生成AI(Generative AI)の医療応用も急速に進展し、患者とのコミュニケーションや医療記録の自動生成など、新たな活用シーンが広がっています。
ReAlice株式会社 開発担当者AIの導入により、医療機器業界では「試行錯誤型」から「予測・最適化型」への開発スタイル転換が進んでいます。モデリングやシミュレーションによる前倒し設計が可能となり、規制対応でも自然言語処理(NLP)を活用した申請書類の自動生成や分類支援が実用化し始めています。
FDA開発AIに関してよくある質問
AI医療機器の承認にはどの程度の期間が必要ですか?
AI医療機器の承認期間は申請経路によって大きく異なります。
510(k)申請では法定審査期間が90日と定められています。事前相談(Pre-Submission)を活用することで、申請前に規制要件を明確化し、効率的な審査プロセスを実現できます。
- 事前相談(Pre-Submission)では60-75日でフィードバック提供
- 法定期間内での審査実現(510(k)は90日)
- 申請時のリスク軽減と遅延防止
- 規制要件の事前明確化
重要なのは、事前相談(Pre-Submission)を活用することで、申請前に規制要件を明確化し、承認期間の短縮を図ることです。FDAは完全な提出パッケージを受領してから60-75日以内にフィードバックを提供することを目指しており、Pre-Submissionプログラムを使用すると510(k)申請はほぼ常に法定期間内で審査されます。
既存の医療機器にAI機能を追加する場合の申請要件は?
既存医療機器へのAI機能追加は、変更の程度により申請要件が決定されます。軽微な変更の場合は特別コントロール(Special Controls)の範囲内で対応可能ですが、診断精度や治療方針に影響する重要な変更の場合は新たな510(k)申請が必要です。
事前変更管理計画(PCCP)を適切に策定していれば、予定された範囲内でのAIモデル更新は追加承認なしで実施できます。
- 軽微な変更:特別コントロール範囲内で対応
- 重要な変更:新たな510(k)申請が必要
- PCCP策定により継続的改善が可能
- Hologic社のGenius™システムが成功例
Hologic社のGenius™システムでは、PCCPを活用してリアルワールドデータに基づく継続的改善を実現しています。
FDAとの事前相談(Pre-Submission)はどのタイミングで行うべきですか?
事前相談は開発の早期段階、特に臨床試験デザインの確定前に実施することが最も効果的です。AI医療機器の場合、学習データセットの設計、アルゴリズムの検証方法、臨床評価の枠組みについて事前に合意を得ることが重要です。
2025年のFDA内部AI導入により、事前相談の回答期間も短縮されており、通常60日程度で回答を得ることができます。
臨床試験デザインの確定前に実施することで、後の変更コストを最小化できます。学習データセットの設計段階で相談することが重要です。
複雑なAI技術を使用する場合は、複数回の事前相談を通じて段階的に合意形成を図ることが推奨されます。回答期間は通常60日程度です。
AI医療機器の市販後変更管理で注意すべき点は?
AI医療機器の市販後変更管理では、継続的学習による性能変化の監視が最重要課題です。
2025年のサイバーセキュリティ脅威予測によると、AIを駆使したより巧妙なフィッシング攻撃やリアルタイムでマルウェアを進化させる攻撃が増加しており、アルゴリズムドリフトやデータポイズニング攻撃への対策として、定期的な性能評価と異常検知システムの実装が必要です。
- AI診断システムへの敵対的攻撃による誤診リスク
- データポイズニングによる学習データ汚染
- ディープフェイクを悪用した医療記録操作
- IoT機器数が320億台に達する予測でのセキュリティ強化
Censinet CEOのEd Gaudetが2025年5月28日に警告したように、AI診断システムへの新興サイバー脅威が患者の生命を直接脅かす時代に入っており、モデル操作による悪性腫瘍の良性誤診や訓練データへのデータポイズニングなどの脅威が現実化しています。また、リアルワールドデータに基づく性能改善を実施する場合は、事前に策定したPCCPの範囲内で実施し、重大な変更の場合は追加承認を取得する必要があります。
日本企業が米国でAI医療機器を販売する際の必要な許可は?
日本企業が米国でAI医療機器を販売するには、FDA承認に加えて施設登録(Establishment Registration)と機器リスト(Device Listing)の提出が必要です。また、米国代理人(US Agent)の指定と品質システム規制(QSR)への適合も義務付けられています。
2023年4月に日本で改定された医療機器基本要件基準のサイバーセキュリティ対策は、FDAの要件とも整合性があるため、日本企業にとって有利な条件となっています。
- FDA承認(510(k)、De Novo、PMA)
- 施設登録(Establishment Registration)
- 機器リスト(Device Listing)の提出
- 米国代理人(US Agent)の指定
- 品質システム規制(QSR)への適合
- 医療機器固有識別子(UDI)システムへの登録
- 有害事象報告(MDR)体制の構築
さらに、医療機器固有識別子(UDI)システムへの登録と、有害事象報告(MDR)体制の構築も必要です。
2025年1月開始予定のジェトロ「HealthTech Gateway “AI Medical in the US”」プログラムでは、Mayo Clinic Platformと連携し、日本のデジタルヘルススタートアップ16社の米国市場参入を支援しており、日本企業にとって有利な環境が整備されています。特に、IEC 81001-5-1規格に準拠したサイバーセキュリティ対策の実装経験は、FDAの厳格なセキュリティ要件への対応において大きなアドバンテージとなることです。
